空间转录组分析工具-seurat介绍
什么是空间转录组(ST)?
以市面上最流行的10X为例:将冰冻组织切片(FF)或石蜡切片(FFPE)放置在10X Genomics Visium芯片的捕获区域内,进行HE染色和成像后,对组织切片进行透化处理,细胞内的mRNA释放出来,从而被芯片上带有oligo-dT的探针捕获,并且每个探针都带有特异的位置序列,然后以mRNA为模版进行cDNA合成,构建文库后再通过测序,获得基因表达信息的同时,每一条测序reads因带有位置序列,从而能够获得基因表达的位置信息。
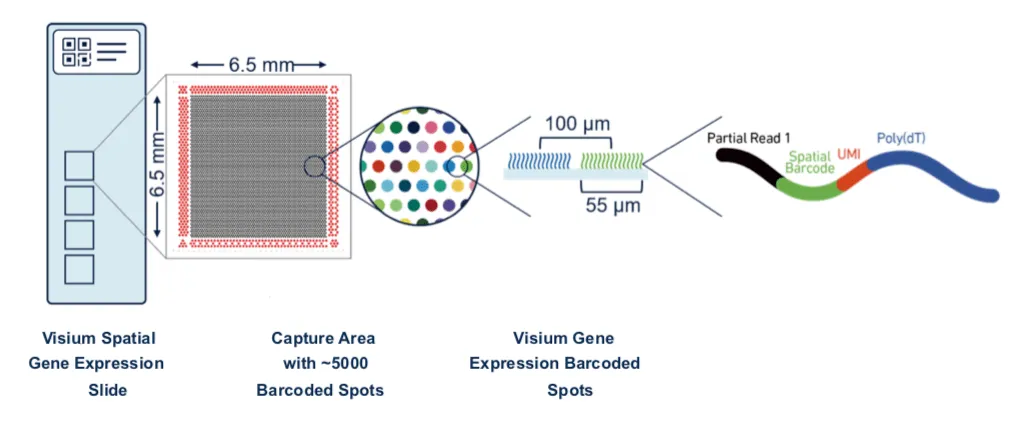
空间转录组的优势
与scRNA-seq相比的优缺点:scRNA-seq将基因表达与单个细胞相关联,但关于这些基因在组织中的位置信息丢失了;相反的,ST知道基因表达的位置,却不知道是哪个细胞产生的。scRNA-seq联合ST能起到1+1>2的作用(超高分辨率+位置信息)。
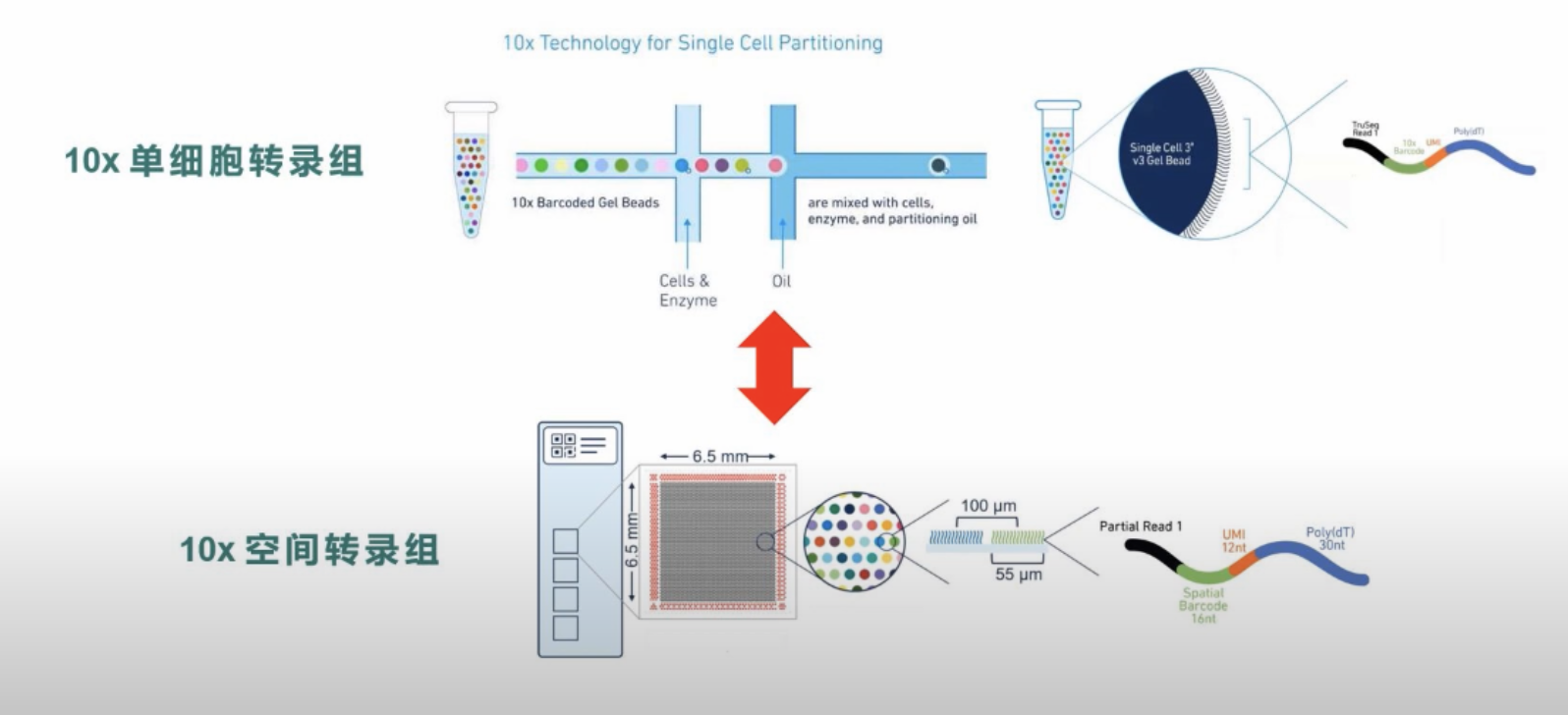
Seurat单细胞空间转录组数据分析
Seurat是一个用于分析单细胞转录组数据的R包,它可以用于对单细胞空间转录组数据进行整合、分析和可视化。Seurat提供了丰富的功能,包括数据预处理、细胞聚类、细胞类型标注、基因差异表达分析等,是单细胞转录组和空间转录组数据分析中常用的工具之一。
Seurat数据分析通常包括以下步骤:
-
数据预处理:
-
读取原始数据,包括单细胞RNA测序数据和空间信息数据。 -
数据质量控制,包括过滤低质量细胞和基因。 -
数据归一化,通常使用对数转换或归一化方法。
-
-
细胞聚类:
-
使用细胞特征进行细胞聚类,常用的方法包括PCA和t-SNE。 -
根据聚类结果对细胞进行分类和标记。
-
-
空间信息整合:
-
将单细胞RNA测序数据与空间信息数据进行整合,以确定细胞在组织空间中的位置。
-
-
空间转录组分析:
-
分析细胞在组织空间中的基因表达模式,探索不同区域的基因表达差异。
-
-
结果可视化:
-
将分析结果可视化,包括细胞聚类图、空间分布图和基因表达热图等,以便进行结果解释和展示。
-
Seurat分析代码实践:
1. 数据预处理:
1.1 导入空转示例数据文件:小鼠大脑组织样本空间转录组数据brain_st
===================================================================
# 提起安装好用到的包,并导入
library(Seurat)
library(SeuratData)
library(ggplot2)
library(patchwork)
library(dplyr)
# 导入空转示例数据文件:a large seurat object
brain <- LoadData("stxBrain", type = "anterior1")
===================================================================
1.2 SCTransform()实现数据归一化
====================================================================
# n count
plot1 <- VlnPlot(brain, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()
plot2 <- SpatialFeaturePlot(brain, features = "nCount_Spatial", slot='data') + theme(legend.position = "right")
wrap_plots(plot1, plot2)
# normalization
brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE)
DefaultAssay(brain)
===================================================================
1.3 特定基因可视化
===================================================================
SpatialFeaturePlot(brain, features = c("Hpca", "Ttr")) # + theme(legend.position = "right")
p1 <- SpatialFeaturePlot(brain, features = "Ttr", pt.size.factor = 3)
p2 <- SpatialFeaturePlot(brain, features = "Ttr", alpha = c(0.1, 1))
p1 + p2
===================================================================
- output:
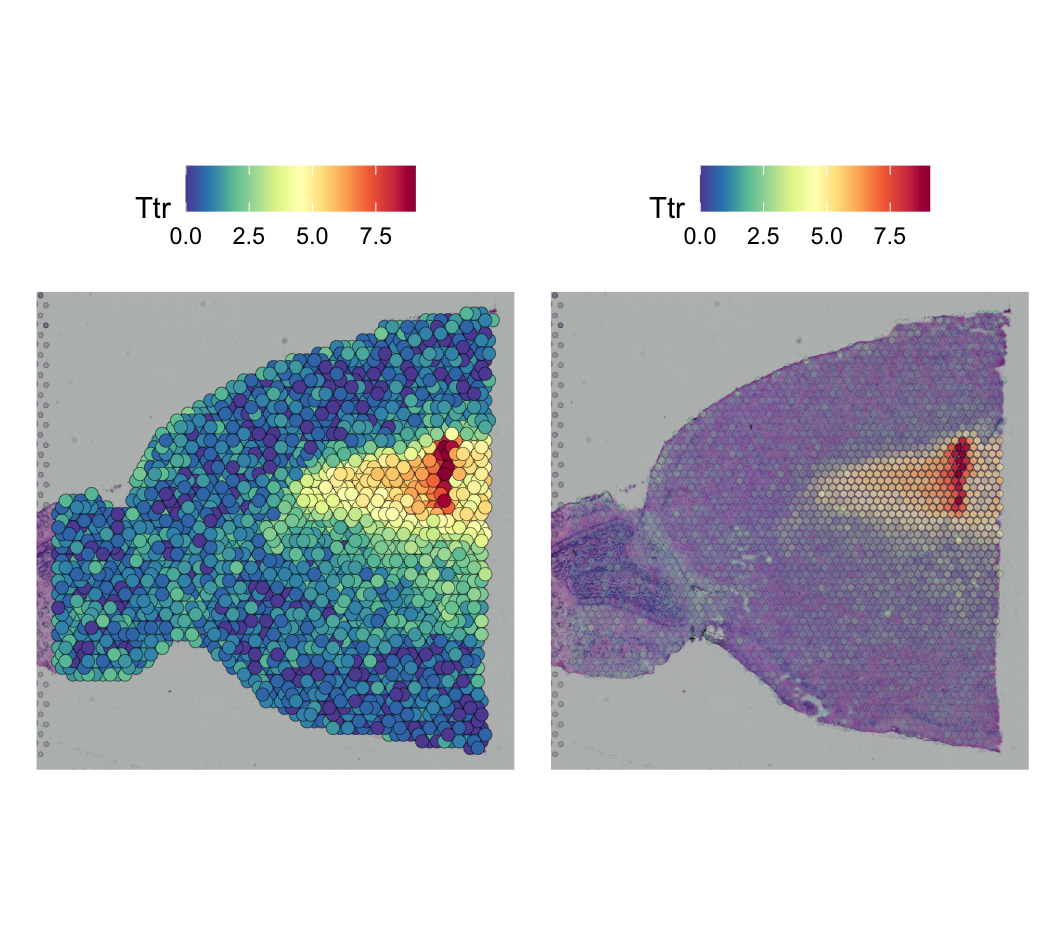
2. 细胞聚类
2.1 降维聚类
-
RunPCA() & RunUMAP()实现降维, FindClusters()实现聚类
===================================================================
brain <- RunPCA(brain, assay = "SCT", verbose = FALSE)
brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30)
brain <- FindClusters(brain, verbose = FALSE)
brain <- RunUMAP(brain, reduction = "pca", dims = 1:30)
p1 <- DimPlot(brain, reduction = "umap", label = TRUE)
p2 <- SpatialDimPlot(brain, label = TRUE, label.size = 3, group.by = 'seurat_clusters')
p1 + p2
===================================================================
-
output: 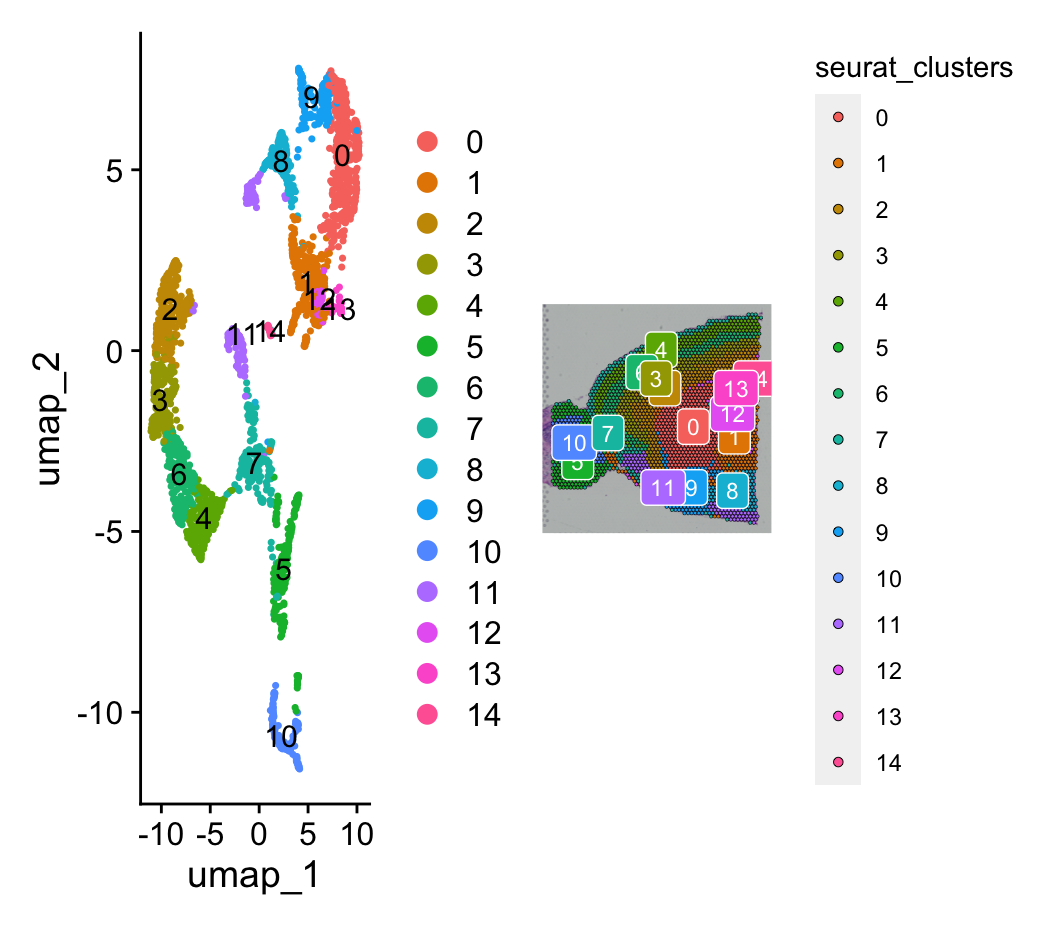
2.2 不同亚群在空间上的单独分布
===================================================================
SpatialDimPlot(brain, cells.highlight = CellsByIdentities(object = brain,
idents = c(2, 1, 4, 3, 5, 8)),
facet.highlight = TRUE, ncol = 3)
===================================================================
- output:
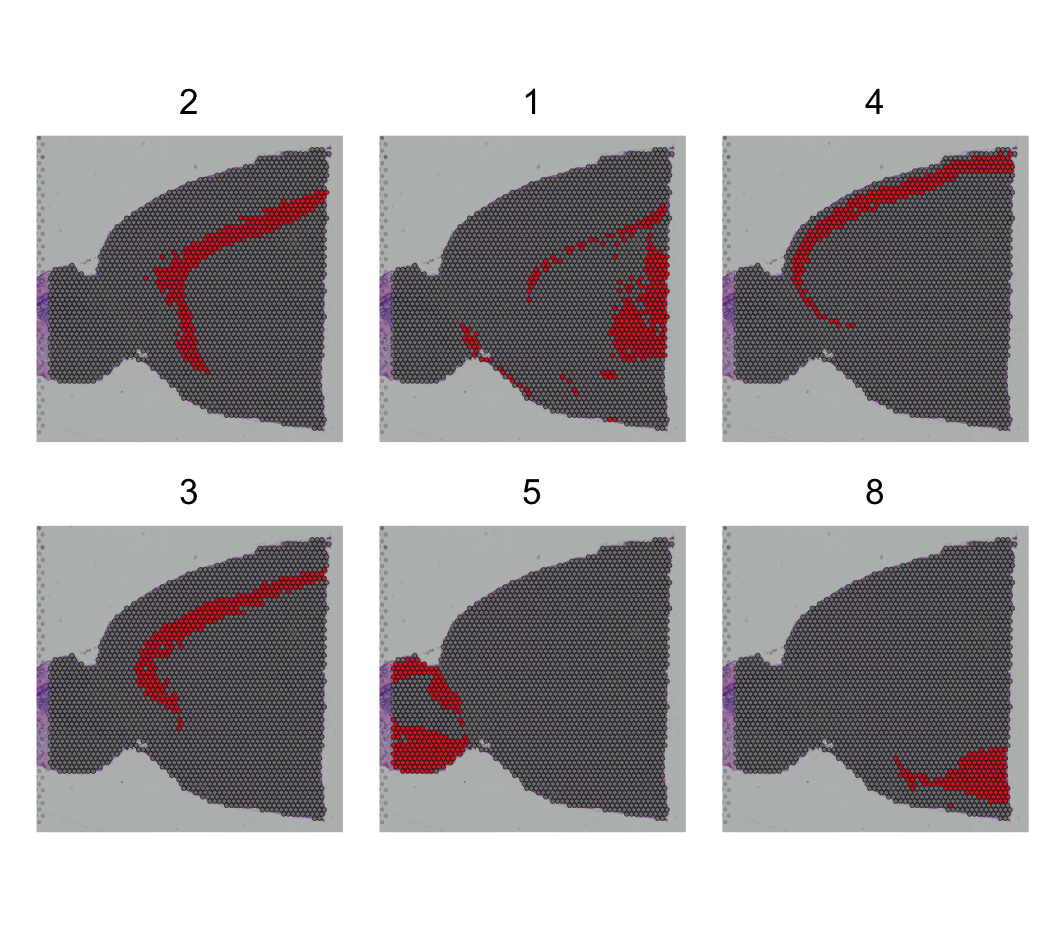
3. 与单细胞RNA-seq数据整合分析,并完成细胞标注
3.1 单细胞转录组数据allen_cortex整合
===================================================================
# 细分解剖区域(截取cortex区域)
cortex <- subset(brain, idents = c(1, 2, 3, 4, 6, 7))
cortex <- subset(cortex, anterior1_imagerow > 400 | anterior1_imagecol < 150, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 275 & anterior1_imagecol > 370, invert = TRUE)
cortex <- subset(cortex, anterior1_imagerow > 250 & anterior1_imagecol > 440, invert = TRUE)
SpatialDimPlot(cortex, crop = T, label = TRUE)
# 与单细胞数据整合
allen_reference <- readRDS("./data/allen_cortex.rds")
allen_reference <- SCTransform(allen_reference, ncells = 3000, verbose = FALSE) %>%
RunPCA(verbose = FALSE) %>%
RunUMAP(dims = 1:30)
# scRNA-seq的细胞注释图
DimPlot(allen_reference, group.by = "subclass", label = TRUE)
===================================================================
3.2 与单细胞数据集成
-
将单细胞RNA-seq的标签map到空间转录组上,鉴定单细胞的空间分布概率
===================================================================
anchors <- FindTransferAnchors(reference = allen_reference, query = cortex,
reference.assay = 'SCT', query.assay = 'SCT',
normalization.method = "SCT", verbose = T, reduction = 'cca')
predictions.assay <- TransferData(anchorset = anchors, refdata = allen_reference$subclass, prediction.assay = TRUE,
weight.reduction = cortex[["pca"]], dims = 1:30)
# 获得每个聚类在每个Spots的预测分数
cortex[["predictions"]] <- predictions.assay
# 基于这些预测分数,预测空间的细胞类型
DefaultAssay(cortex) <- "predictions"
SpatialFeaturePlot(cortex, features = c("Astro", "L2/3 IT"), pt.size.factor = 1, ncol = 2, crop = FALSE, alpha = c(0.1, 1))
===================================================================
4. 多切片,多样本分析
===================================================================
# merge多样本
DefaultAssay(brain) <- 'SCT'
brain2 <- LoadData("stxBrain", type = "posterior1")
brain2 <- SCTransform(brain2, assay = "Spatial", verbose = FALSE)
brain.merge <- merge(brain, brain2)
# 降维聚类
DefaultAssay(brain.merge) <- "SCT"
VariableFeatures(brain.merge) <- c(VariableFeatures(brain), VariableFeatures(brain2))
brain.merge <- RunPCA(brain.merge, verbose = FALSE)
brain.merge <- FindNeighbors(brain.merge, dims = 1:30)
brain.merge <- FindClusters(brain.merge, verbose = FALSE)
brain.merge <- RunUMAP(brain.merge, dims = 1:30)
# 可视化
DimPlot(brain.merge, reduction = "umap", group.by = c("ident", "orig.ident"))
SpatialDimPlot(brain.merge)
SpatialFeaturePlot(brain.merge, features = c("Hpca", "Plp1"))
SpatialFeaturePlot(brain.merge, features = c("Hpca", "Plp1"), images = 'anterior1')
=======================================================================
请关注微信公众号,更多精彩内容实时更新中

本网站发布所有原创内容,版权归属尖端生物及相关版权方,内容仅供学术交流,如有侵权请联系删除。未经授权的转载是侵权行为,版权方保留追究法律责任的权利。投稿,转载或版权问题,请联系:submit@advanced-biotech.cn;商务合作请联系:cc@advanced-biotech.cn
- 2024-08-15
- 2024-08-13
- 2024-08-05
- 2024-07-24
- 2024-07-23
- 2024-07-22
- 2024-07-18
- 2024-07-10




